

Next Lesson - When Haematopoiesis Goes Wrong
Core
Haemoglobin (Hb) is a globular protein composed of four protein chains. These four chains are made up of 2α paired with 2 other chain types (see table below for normal variants of these chains). Attached to each of globin chain is an iron (Fe2+) ion which allows for oxygen bonding.
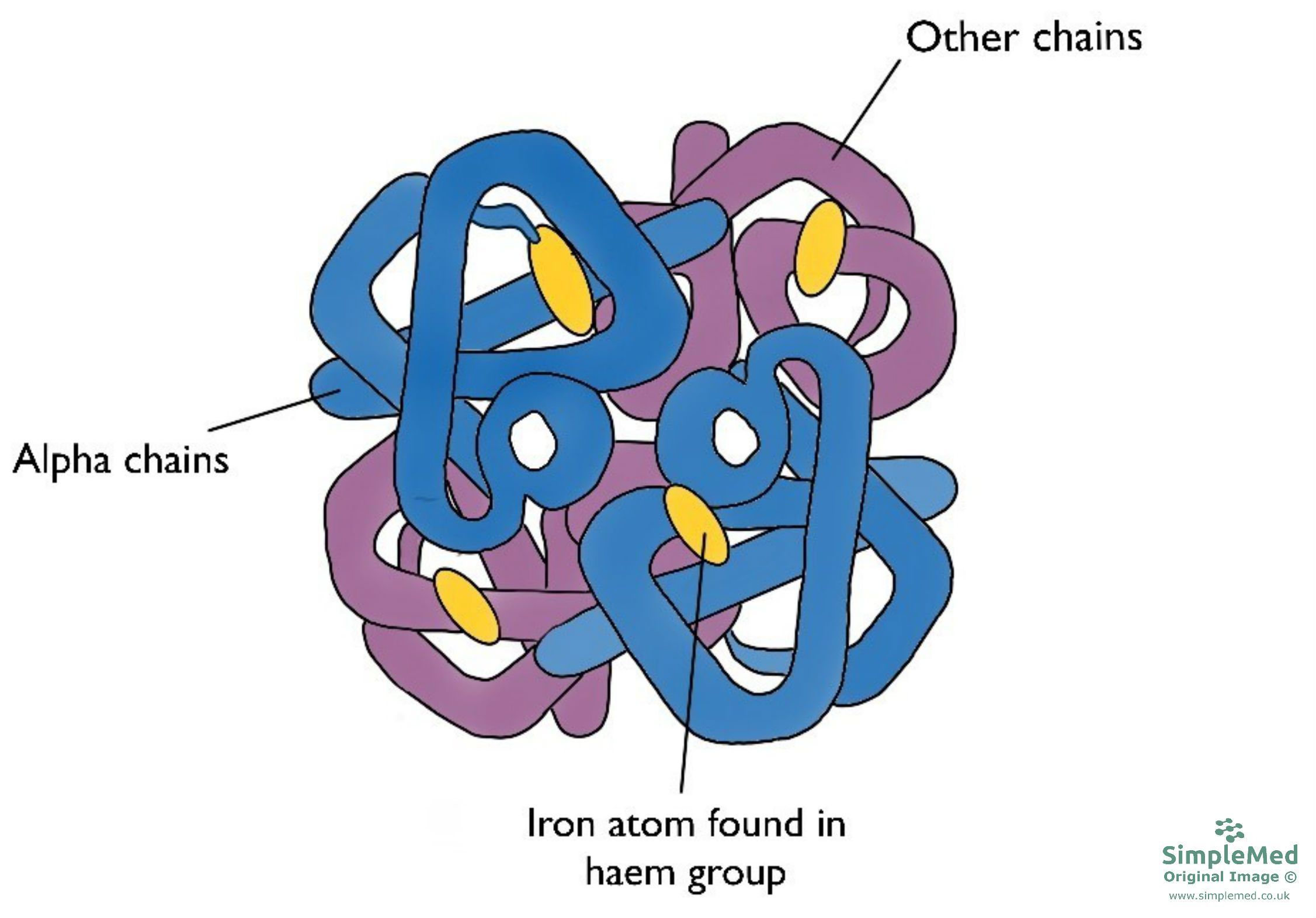
Figure 1. The structure of haemoglobin
SimpleMed original by Dr. Keertana Anne
Common forms of physiological haemoglobin that are found in the adult body are highlighted in the table below.
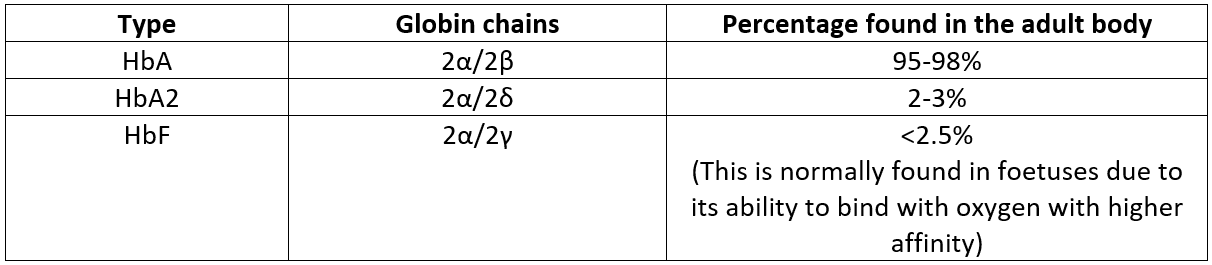
Figure 2. A table of the different haemoglobin variants
SimpleMed original by Dr. Keertana Anne
Mutations to the DNA which codes for Hb can produce haemoglobin variants. Some mutations cause no changes and go undetected, and these are called non-pathological variants. On the other hand, some mutations may be pathological and lead to haemoglobinopathy.
Haemoglobinopathies are genetic disorders (usually autosomal recessive) that alter the structure of haemoglobin. This may result in deformed structures, or reduced production of particular globin chains - called thalassaemias (covered in more detail later). This results in reduced oxygen carrying capacity of the blood, and produces symptoms of anaemia (for more details, see our article on Anaemia).
The most common types of haemoglobinopathies are thalassaemias and sickle cell disease.
Thalassaemias are autosomal recessive, inherited diseases resulting from the reduced rate of synthesis of normal α- or β- globin chains. This results in a lower level of intracellular haemoglobin which causes hypochromic, microcytic red cells seen in patients with thalassaemia. In thalassaemia, abnormal red cells are broken down in the spleen, contributing to anaemia. Iron from haem is normally recycled, but patients may develop iron overload due to increased intestinal absorption and repeated blood transfusions. Splenomegaly and hypersplenism can worsen anaemia, but splenectomy is reserved for selected cases because of its long-term risks.
Thalassaemias are classified based on which globin chain is affected. α-globin chains are encoded by two closely linked genes on chromosome 16 (so therefore four loci), while β-globin chains are encoded by a single gene on chromosome 11 (therefore two loci).
α-thalassaemia:

Figure 3. Chromosome 16 with the location of the α-globin chain gene shown
SimpleMed original by Dr. Keertana Anne
α-thalassaemias are caused by mutations (usually deletions) in the HBA1 and/or HBA2 genes on chromosome 16. The increased deletion of α chains means there are an excess of β chains in adults, and γ chains in fetuses. This means the patient is unable to form the normal globular proteins in the pattern of 2α and 2 other. The relative excess of these other chains leads to defective red cells, which are destroyed in large numbers by the spleen - this causes haemolytic anaemia.
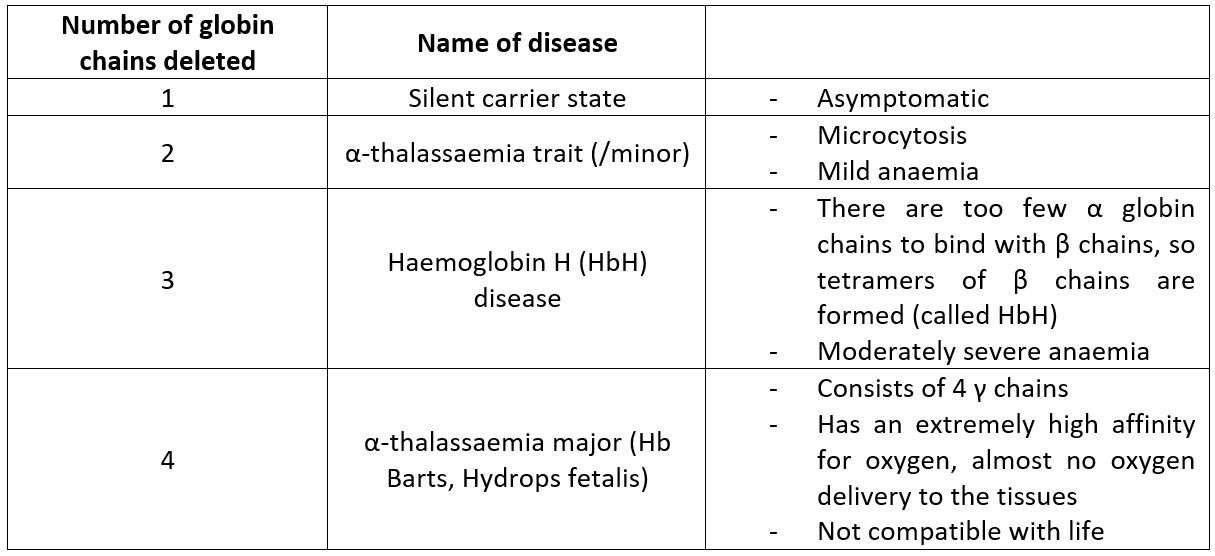
Figure 4. A table of the different types of α-thalassaemias
SimpleMed original by Dr. Keertana Anne
Alpha thalassaemias are most commonly seen in sub-Saharan African, southeast Asian, Middle Eastern and Mediterranean populations.
β-thalassaemia:
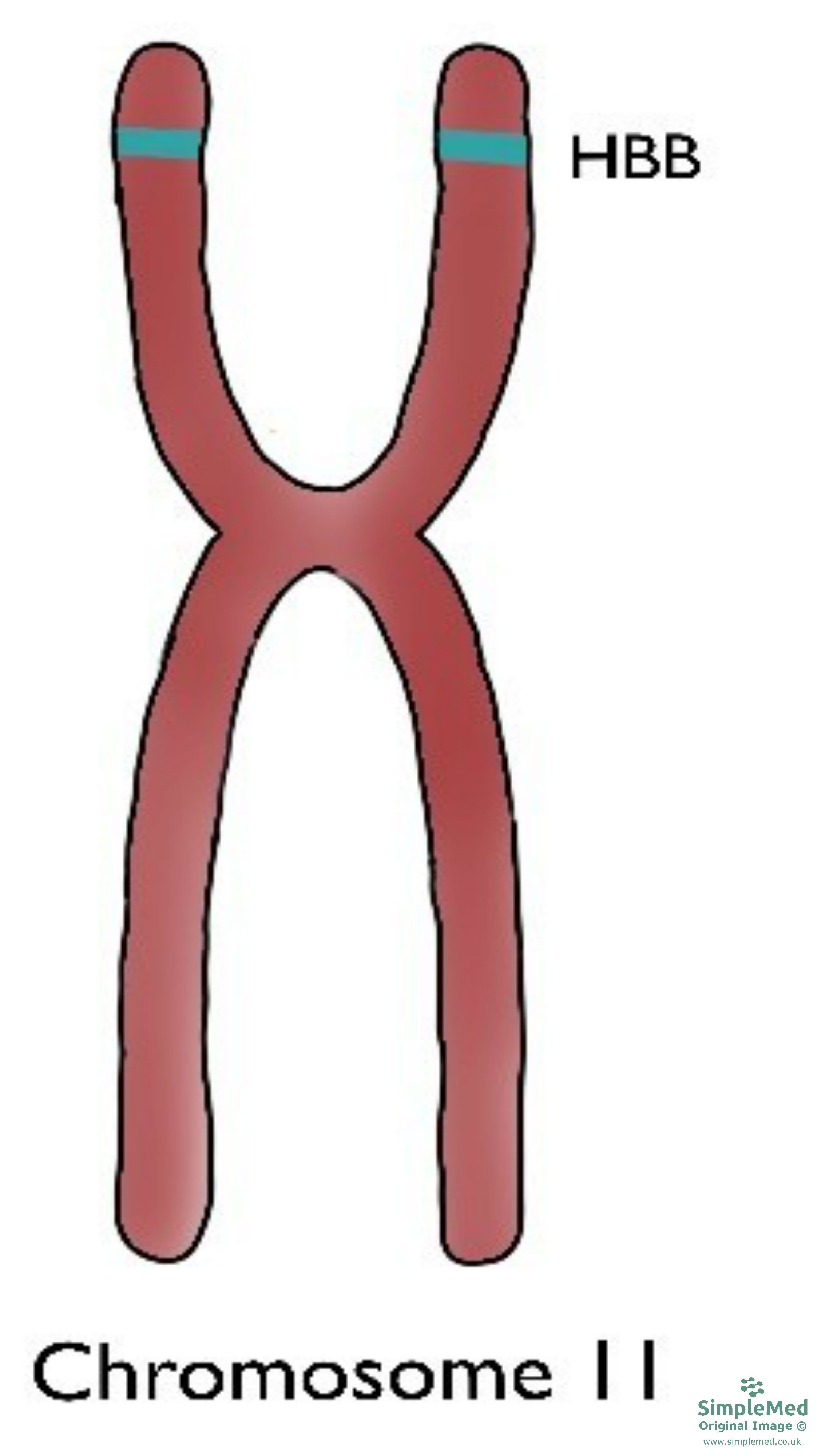
Figure 5. Chromosome 11 with the location of the β-globin chain gene shown
SimpleMed original by Dr. Keertana Anne
Beta thalassaemias are caused by mutations in the HBB gene on chromosome 11. The body’s inability to construct β chains leads to the underproduction of HbA - which is the most prominent haemoglobin variant in the body. This, similarly to α-thalassaemia, leads to microcytic anaemia.
There are 3 main types of β-thalassaemia: thalassaemia minor, thalassaemia intermedia and thalassaemia major.
- Thalassaemia Minor
- This is the form of β-thalassaemia where only one of the alleles in chromosome 11 has a mutation, this patient is a carrier.
- The patient doesn't exhibit severe symptoms, but will have mild microcytic anaemia.
- Patients are often monitored without treatment (unnecessary transfusions may result in iron overload, leading to transfusion haemosiderosis causing damage to the liver, heart and endocrine glands).
- Thalassaemia Intermedia
- In patients with beta thalassaemia intermedia, anaemia is present (symptoms range from mild to severe) but individuals are not transfusion dependent.
- Patients are clinically heterogeneous.
- Thalassaemia Major
- Also known as Cooley’s anaemia, this is the form where both alleles in chromosome 11 have mutations (homozygous recessive).
- Due to the massively reduced ability to produce HbA, the patient has severe microcytic and hypochromic anaemia and is transfusion and chelation (to treat iron overload) dependent.
- Patients may have abnormal skull bones due to excessive extramedullary haemopoiesis in an attempt to keep up with haemolysis.
- Hepatosplenomegaly is also present as a result.
Sickle Cell Disease and Sickle Cell Anaemia
Sickle cell disease (SCD) is another inherited haemoglobinopathy. Normally red blood cells have a biconcave shape and are flexible, meaning they can easily move through blood vessels.
However, in sickle cell disease, when deoxygenated the defective haemoglobin S (HbS - haemoglobin specific to sickle cell disease) polymerises causing deformation of the cell, losing its biconcave structure and becoming ‘sickle’ (or crescent moon) shaped. This irregular shape causes the sickle cells to become stuck in blood vessels, impairing the flow of blood to certain areas of the body. Furthermore, the cells are quite fragile so easily break down or are removed by the spleen, leading to a haemolytic anaemia.

Figure 6. Image showing the differences between normal red blood cells and sickle cells in blood vessels
SimpleMed original by Dr. Keertana Anne
Like thalassaemia, sickle cell disease is inherited in an autosomal recessive pattern. In sickle cell disease, abnormal haemoglobin S (HbS) is produced due to a mutation in the beta globin gene; the most severe form occurs when both beta chains are HbS, leading to marked sickling of red cells. Sickle cell anaemia, a severe type of SCD, is where both β globin chains in HbA are replaced with HbS (homozygous recessive). Sickle cell trait can be found in up to 30% of the West African population, as it has been shown to be protective against malaria (sickle cell disease does not protect against malaria).
Signs and symptoms of sickle cell disease usually begin in early childhood and are caused by the sickle shape of RBC. The premature breakdown of the cells leads to anaemia (which can cause dyspnoea, fatigue and tachycardia) and if severe enough, pre-hepatic jaundice. If sickle cells get stuck in blood vessels this can lead to vaso-occlusive crises (which are very painful), as tissues and organs are deprived of oxygen and become ischaemic. One of the most severe complications of sickle cell anaemia is acute chest syndrome, which is a serious condition and is also exquisitely painful.
Although treatments are available to help manage the condition and alleviate symptoms of the disease, the only curative treatment is a bone marrow transplant. Due to hyposplenism caused by multiple infarcts, prophylactic antibiotics and immunisation should be given. Despite advancing care, patients have a greatly reduced life expectancy (67 years in the UK), the most common causes of death being strokes, multi-organ failure and acute chest syndrome.
Edited by: Dr. Thomas Burnell and Dr. Ben Appleby
Reviewed by: Dr. Marcus Judge
In this article
Haemoglobinopathies are autosomal recessive disorders affecting haemoglobin. Thalassaemias, which involve reduced production of globin chains rather than…
- 10900
