

Next Lesson - Haematology in Systemic Disease
Core
To better understand this topic, have a read of our article on Haematopoiesis and the Spleen.
As previously mentioned, haematopoiesis is the formation of the cellular components of blood. A healthy adult may produce an average of around 200,000,000,000 red cells, approximately 1,000,000,000 to 10,000,000,000 white cells, and about 100,000,000,000 platelets per day.
A deviation from the norm (i.e. too many or too few cells are produced) can produce life threatening complications. This deviation can be caused by infections, certain medications or malignancy.
The overproduction of blood cells has two main causes; damage to the bone marrow or abnormalities in hormonal signalling.
Overproduction due to diseases in the bone marrow are called myeloproliferative neoplasms. These arise from genetic mutations in the common myeloid progenitor cells, and increase levels of various blood cells including platelets, erythrocytes and white blood cells in the body.
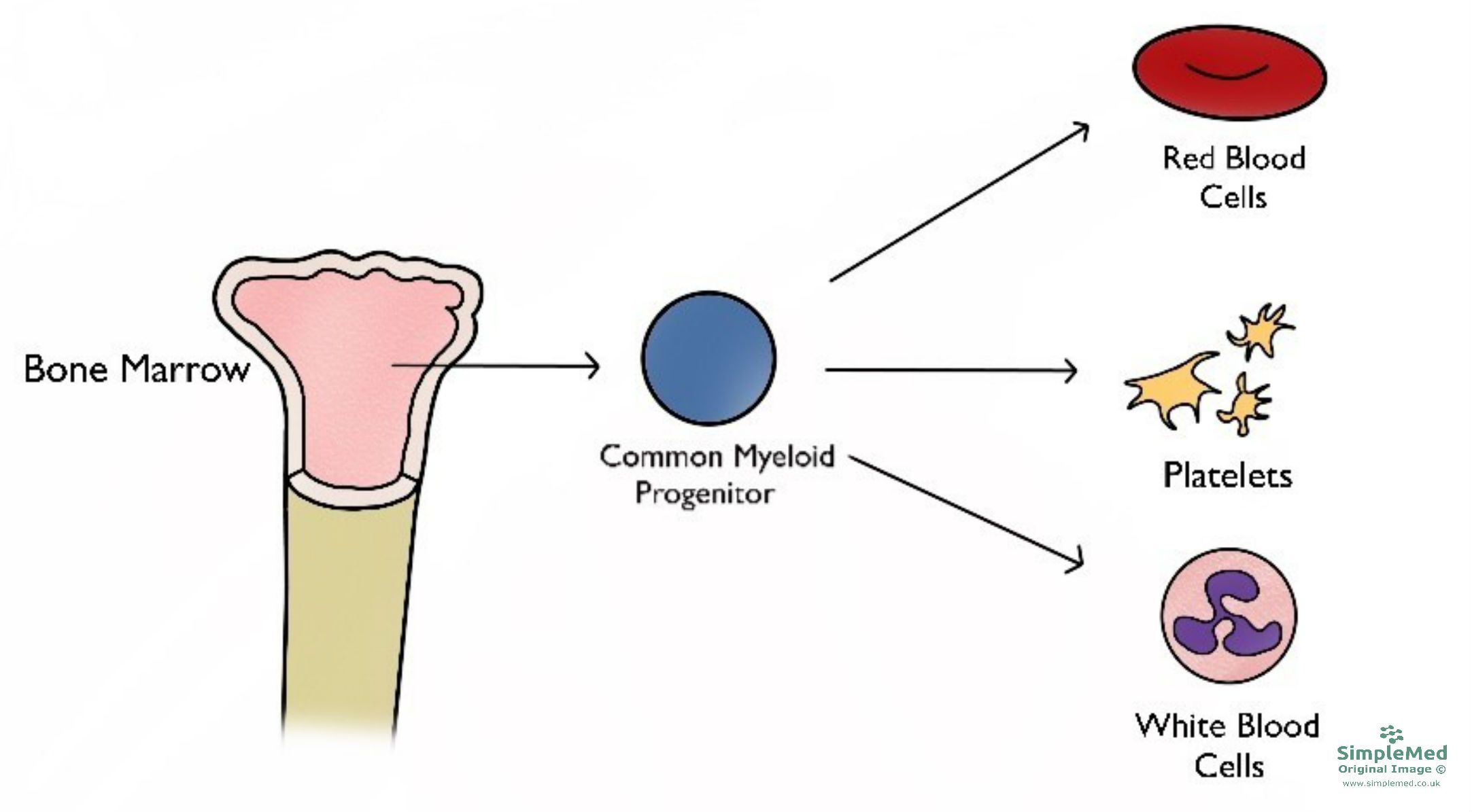 Figure 1 - The types of cells that arise from the bone marrow, specifically the myeloid lineage
Figure 1 - The types of cells that arise from the bone marrow, specifically the myeloid lineage
SimpleMed original by Dr. Keertana Anne
Polycythaemia is a disease state where the haematocrit is raised (greater than 52% in males and over 48% in females).
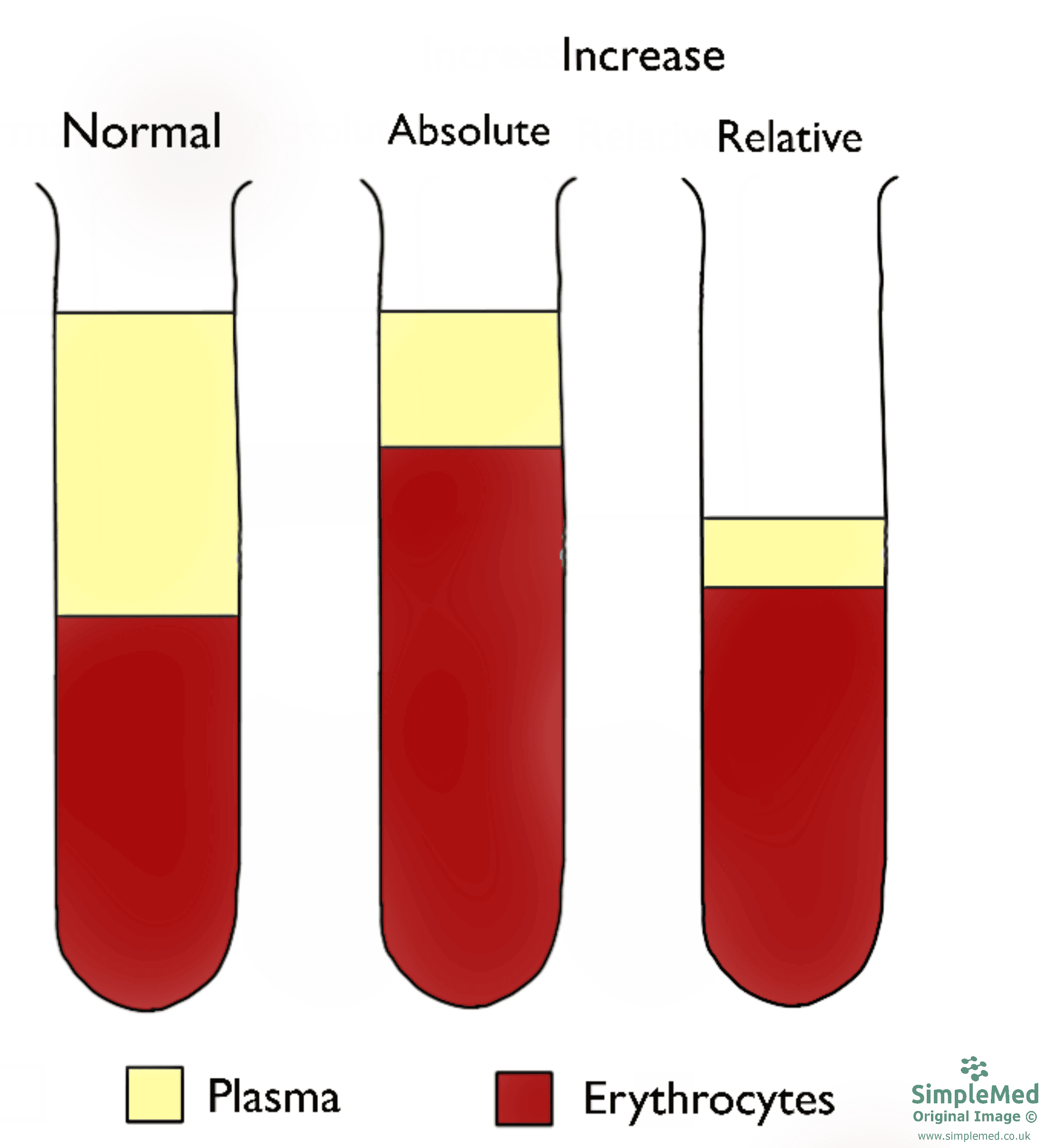
Figure 2 - The difference between an absolute and a relative increase in the haematocrit level
SimpleMed original by Dr. Keertana Anne
An absolute increase in haematocrit is due to increased erythrocytes, while a relative increase is due to a decrease in plasma volume (note how volume of erythrocytes remains the same).
An increase in erythrocytes (absolute increase in haematocrit) can happen for a number of reasons:
- Primary causes - When there is an abnormality in the bone marrow, known as polycythaemia vera.
- Secondary causes - There are increased levels of erythropoietin:
- Physiological due to increased oxygen requirement, e.g. high altitude or chronic lung conditions.
- Pathological due to abnormal production from the kidneys or from a secreting tumour (e.g. renal cell carcinoma or hepatocellular carcinoma).
- Self administered human erythropoietin (EPO) is used by some members of the sporting community to increase performance. EPO is banned by the world anti-doping association (WADA).
The most common cause for a raised haematocrit however is a relative increase. This is usually due to dehydration, which can be resolved by replenishing fluids.
Polycythaemia vera is a specific type of polycythaemia, which is a myeloproliferative neoplasm (bone marrow disorder). These are usually caused by mutations in the gene coding for the enzyme Janus Kinase 2 (JAK2), leading to abnormal signalling within bone marrow cells that drives excessive blood cell production, even in the absence of normal regulatory signals. Alternatively, extra red blood cells may have been received accidentally through transfusion, or due to delayed clamping of the umbilical cord after delivery. This mainly leads to an increase in erythrocyte production, but can also increase levels of white blood cells and platelets.
The signs and symptoms of polycythaemia are generally non specific and may include dizziness, headaches and tinnitus. Pruritis (itching) after a warm bath and erythromelalgia (a burning sensation in the fingers and toes with associated erythema) are characteristic. Many cases will be asymptomatic.
Overproduction of red blood cells is treated by therapeutic phlebotomy (bleeding the patient), which reduces haematocrit levels. Drugs that inhibit production can be used if this is not tolerated.
When presented with a high haematocrit it is helpful to first consider more common causes. Therefore first consider, is this relative change to the haematocrit, and if not is this a secondary cause. A primary cause (being the least common can then be considered.
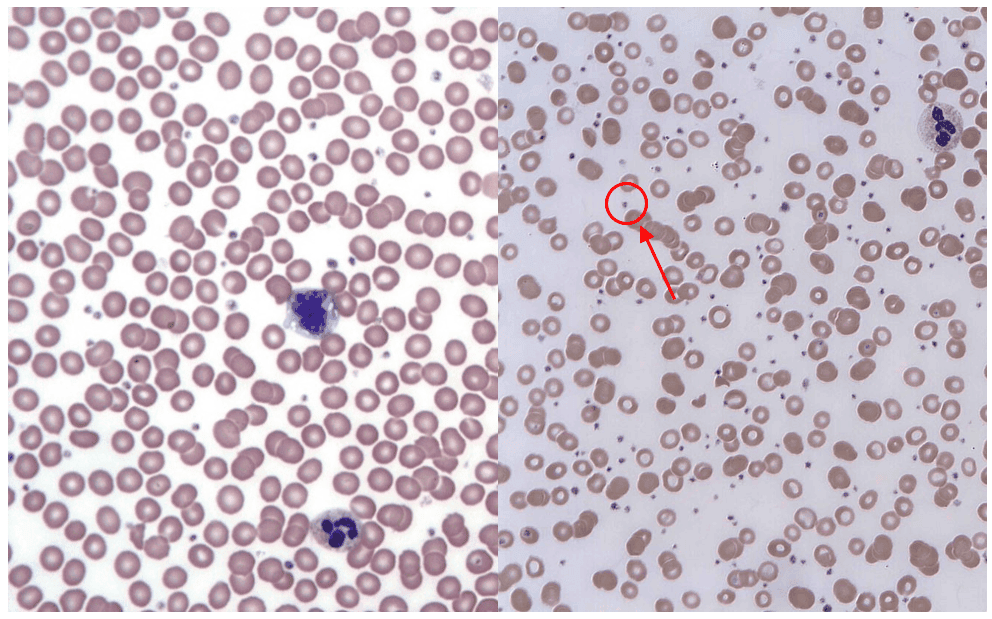
Figure 3 - A normal blood film vs the blood film of a patient with thrombocytosis. The small dots (circled) are platelets
Creative commons source by Keith Chambers [CC BY-SA 4.0 (https://creativecommons.org/licenses/by-sa/4.0)] - Left Image
Creative commons source by Erhabor Osaro (Associate Professor), edited by Dr. Keertana Anne [CC BY-SA 4.0 (https://creativecommons.org/licenses/by-sa/4.0)] - Right Image
Thrombocytosis is an increase in the platelet count of the blood relative to that patient’s age and gender. A physiological increase in platelets is commonly seen in hospitalised patients, due to a higher risk of infection and inflammation. Pathologically there are two causes of thrombocytosis: reactive (secondary) and primary.
Reactive thrombocytosis is the more common pathological cause, and develops secondary to another cause. These causes include chronic inflammatory disorders, acute infections (this risk is increased with spleen removal) and cancers. The function of platelets is usually normal in reactive thrombocytosis, and the thrombotic risk is generally much lower than in primary causes, although it depends on the underlying condition.
Primary thrombocytosis (also known as Essential Thrombocythaemia), on the other hand, is a myeloproliferative neoplasm. This is a rare blood cancer, causing an overproduction of megakaryocytes (the precursor to platelets) that originates in the bone marrow. A JAK2 mutation is seen in about 50% of these patients, while in others a mutation in the thrombopoietin receptor is seen (this regulates megakaryocyte and therefore platelet production). This produces a functional disorder, and the symptoms relate to an increase in platelets. Symptoms include increased thrombosis risk, numbness in the extremities and easy bruising.
Leukaemia is a group of blood cancers that originate in the bone marrow, producing increased numbers of abnormal white blood cells that are unable to perform their normal function as they are immature.
Leukaemia is categorised in two ways: whether it is acute or chronic; and whether it is myeloid or lymphoid.
Acute leukaemia results from a rapid increase in the number of immature blood cells, and these overwhelm the ability of the bone marrow to produce mature blood cells. This rapidly produces the symptoms of leukaemia. Chronic leukaemias on the other hand are slower to produce the symptoms (may take months to years), as the blood cells that are produced are relatively mature, but still abnormal.
Myeloid leukaemias originate from the myeloid lineage, and so affect erythrocytes, granulocytes and platelets. Lymphoid (lymphocytic) leukaemias originate from the lymphoid lineage, and so affect lymphocytes and natural killer cells.
It is important to remember that lymphoid leukaemia’s are distinct from lymphomas (which are classified into Hodgkin and non-hodgkin) due to their location. Although both are caused by issues with precursor cells, in leukaemia the cancer cells mainly reside in the bone marrow and blood whereas lymphoma cells reside in lymph nodes and other tissues.
Frequent infections are a key complication of leukaemias. This is because the immature white blood cells are no longer able to remove pathogens, meaning previously common infections can become life-threatening. Other signs and symptoms come from abnormal platelets (the clotting process is dysfunctional, leading to increased bleeding and bruising) and abnormal erythrocytes (the immature red blood cells can no longer carry enough oxygen to tissues, causing anaemia - see our article on anaemia).
| Acute | Chronic |
Myeloid | Acute Myeloid Leukaemia | Chronic Myeloid Leukaemia |
Lymphoid | Acute Lymphoblastic Leukaemia | Chronic Lymphocytic Leukaemia |
Insufficient numbers of blood cells can also be produced due to damaged bone marrow, and these changes can be either benign or malignant.
Thrombocytopenia is characterised by an abnormally low level of platelets, the causes of which can either be inherited or acquired. The lack of platelets means that the body is no longer able to form blood clots easily which increases the risk of bleeding greatly.
There are many different types of inherited thrombocytopenias, and all of them are very rare. Some examples include Alport syndrome, May-Hegglin anomaly and Fanconi anaemia. These are easily misdiagnosed due to their rarity.
Acquired thrombocytopenias are a lot more common. Patients with these disorders experience symptoms such as bleeding gums, nosebleeds and petechiae (small bruises on the skin) on their feet, shins and forearms. Women may experience heavier and longer menstrual periods. The following three things can cause acquired thrombocytopenia:
- Decreased platelet production: lack of vitamin B12 or folate (unable to form megakaryocytes); liver failure (decreased production of thrombopoietin).
- Increased platelet consumption: massive haemorrhage; disseminated intravascular coagulation (condition where blood clots form throughout the body, blocking small vessels and using up platelets); thrombotic thrombocytopenic purpura.
- Increased platelet destruction: hypersplenism (there is a greater destruction of platelets and increased splenic pooling of platelets, thereby removing them from circulation); drug induced; immune thrombocytopenic purpura.
Thrombotic Thrombocytopenic Purpura (TTP)
TTP is a blood disorder characterised by widespread microthrombi forming in small blood vessels, leading to a reduced platelet count. Although it shares some features with disseminated intravascular coagulation (DIC), TTP is caused by a severe deficiency of ADAMTS13, resulting in the accumulation of ultra large von Willebrand factor multimers and inappropriate platelet aggregation, rather than thrombin excess.
TTP is associated with the use of certain drugs and chemotherapy, whereas DIC is triggered by head trauma or sepsis. Blood films in TTP typically show an increased number of schistocytes, while DIC may also show schistocytes, although this finding can be variable.
Immune Thrombocytopenic Purpura (ITP)
ITP is an autoimmune disease that is caused by anti-platelet auto-antibodies and T-cell activity against platelets and megakaryocytes. This can be triggered by infections and other associated autoimmune diseases, such as rheumatoid arthritis or systemic lupus erythematosus, however, the cause is unknown in 80% of the cases.
The symptoms of ITP are due to the low platelet count, e.g. excessive bruising, bleeding gums, etc. ITP is usually treated with immunosuppressive therapies, most commonly corticosteroids, depending on the severity of thrombocytopenia and bleeding risk.
Myelodysplastic syndromes (MDS) are a rare group of cancers that affect the blood-forming cells of the bone marrow. The immature blood cells that are produced are destroyed soon after being released from the bone marrow, and over time, there are more immature and defective cells than healthy ones. Different cell lineages can be affected, however, the most common finding is a shortage of red blood cells (therefore the patient presents with anaemia).
Some MDS are idiopathic (no known cause), while others have factors that contribute to the risk (such as: chemotherapy or radiotherapy; certain chemicals [tobacco, pesticides] and certain heavy metals).
Myelodysplastic syndromes are split into subtypes based on the cell lineage affected: red blood cells, white blood cells or platelets. Many patients don’t exhibit any symptoms, but the signs produced are non-specific and related to the blood cytopenias: anaemia (low red blood cells - fatigue, dyspnoea and tachycardia); neutropenia (low neutrophils - increased infection risk); thrombocytopenia (low platelets - increased bleeding risk).
Although about 1 in 3 patients with myelodysplastic syndromes might eventually develop leukaemia (specifically acute myeloid leukaemia), about 50% of deaths occur as a result of bleeding or infection.
Aplastic anaemia is a rare autoimmune condition in which your body stops producing sufficient new blood cells of all three types (pancytopenia) as a result of bone marrow damage (either aplasia or hypoplasia - a complete or partial reduction in the stem cells available).
Aplastic anaemia is often confused with myelodysplastic syndrome, however, unlike myelodysplastic syndrome, which can affect individual cell lineages, aplastic anaemia causes a deficiency of all blood cell types. The symptoms that are produced, however, are very similar to MDS.
Aplastic anaemia can be caused by immune disorders or exposure to chemicals, radiation or drugs, but about 50% of the cases are idiopathic.
Treating aplastic anaemia involves suppression of the immune system using medication, or in more severe cases, a bone marrow transplant - the latter is usually only given in younger patients. Risks with transplants include ‘graft-vs-host disease’, in which the newly created white blood cells may attack the rest of the body. With treatments, patients have a five-year survival rate that exceeds 45%.
Myelofibrosis is classified as a type of myeloproliferative neoplasm in which the excessive multiplication of abnormal haematopoietic stem cells in the bone marrow causes fibrosis. The abnormal cells produce cytokines, which eventually cause the marrow to be replaced with scar tissue. The decrease in functioning tissue means the patients have a reduced ability to produce new blood cells, and progress to pancytopenia. The cause of this disease is usually associated with JAK2 mutations, but patients can also have mutations in the CALR and MPL genes (only 10% of cases carry none of these mutations).
The progressive replacement of healthy bone marrow with scar tissue causes the progenitor cells to begin to colonise the liver and spleen, leading to extramedullary haematopoiesis (the production of blood cells outside the bone marrow medulla). This can present with hepatomegaly and splenomegaly. Patients usually have symptoms of bruising, fatigue, increased sweating and weight loss.
Myelofibrosis is described as primary if the disease is spontaneously or idiopathic, and secondary if it is a consequence of polycythaemia vera or essential thrombocythaemia.
There is currently no definitive cure for most patients, and prognosis varies depending on disease severity and risk factors. Treatment is largely supportive and may include blood transfusions, symptom control with targeted therapies, and splenectomy in selected cases.
Edited by: Dr. Ben Appleby and Dr. Thomas Burnell
Reviewed by: Dr. Marcus Judge
In this article
A deviation from the normal quantity and quality of blood cells produced can have massive consequences for patients.
- 8621

{kind=link}