

Next Lesson - Pharmacodynamics
Core
Introduction to Pharmacokinetics
Pharmacokinetics, often shortened to PK, is the study of how the body handles a drug from the moment it is administered to the moment the last molecule is removed. A clean way to remember the difference between pharmacokinetics and pharmacodynamics is that pharmacokinetics is what the body does to the drug, and pharmacodynamics is what the drug does to the body.
For a medicine to work, it has to reach its site of action at a sufficient concentration and stay there long enough to exert its effect, without ever rising into the toxic range. Every drug that has ever been licensed has been licensed on the back of a pharmacokinetic profile that allows this to happen reliably in most patients.
The ADME Framework
Almost every question in pharmacokinetics can be filed under one of four headings:
- Absorption: getting the drug into the systemic circulation.
- Distribution: moving it from the blood into the tissues where it acts (and into compartments where it does not).
- Metabolism: chemically modifying it, usually in the liver, to make it easier to excrete.
- Excretion: removing it from the body, predominantly via the kidneys.
These four processes happen simultaneously rather than in sequence, but it is far easier to learn them one at a time.
Diagram: The four processes of pharmacokinetics: absorption, distribution, metabolism and excretion.
Why Pharmacokinetics Matters in Practice
Around 1 in 20 prescriptions issued in the UK contains an error, and the consequences range from minor inconvenience to fatal toxicity. Many of those errors involve pharmacokinetic considerations: a dose too large for the patient's renal function, a drug that interacts with another by inducing a liver enzyme, or a frequency that does not match the half-life. The British National Formulary (BNF) and NICE guidance both lean heavily on pharmacokinetic principles when they recommend particular doses, intervals and monitoring strategies.
Several patient factors routinely change pharmacokinetics and need to be considered before any drug is prescribed:
- Renal function, particularly important for water-soluble drugs cleared by the kidneys (e.g. gentamicin, digoxin, lithium). See Glomerular Filtration Rate and the Measurement of Kidney Function.
- Hepatic function: relevant for drugs that undergo extensive hepatic metabolism (e.g. warfarin, opioids, statins). See Hepatic, Biliary and Pancreatic Pathology.
- Age: neonates and the elderly handle drugs differently to younger adults.
- Pregnancy: fluid balance, plasma protein levels and renal blood flow all change.
- Concomitant medication: many of the most important drug-drug interactions are pharmacokinetic in nature, although pharmacodynamic interactions are also common.
- Acute illness: pyrexia, sepsis and shock can all alter absorption, distribution and clearance.
Absorption
Absorption is the process by which a drug crosses the membranes between its site of administration and the systemic circulation. The route of administration largely determines how completely, and how quickly, this happens. The principles of how molecules cross cell membranes are covered in Biological Membranes.
Routes of Administration
Routes are broadly divided into enteral (via the gastrointestinal tract) and parenteral (everything else).
Enteral routes:
- Oral (PO): the most common route. Convenient, cheap and safe, but absorption is slow, variable and exposed to first-pass metabolism. Most absorption occurs in the small intestine, the anatomy of which is described in Anatomy and Physiology of the Midgut.
- Sublingual / buccal: drug is absorbed across the oral mucosa directly into the systemic circulation, bypassing the liver. Useful for drugs that would otherwise be heavily metabolised on first pass (e.g. glyceryl trinitrate).
- Rectal (PR): absorbed via haemorrhoidal veins; the lower rectum drains directly to systemic circulation, partially bypassing the liver. Useful when a patient cannot swallow or is vomiting (e.g. diclofenac, paracetamol, diazepam).
Parenteral routes:
- Intravenous (IV): drug is delivered straight into the bloodstream, giving a rapid onset and 100% bioavailability.
- Intramuscular (IM) and subcutaneous (SC): slower than IV; absorption depends on local blood flow.
- Inhaled: the large surface area of the lungs allows fast absorption; useful for drugs intended to act locally on the airways (salbutamol) or systemically (volatile anaesthetics).
- Transdermal: patches give slow, sustained release through the skin (fentanyl, oestrogen, nicotine).
- Topical: applied to skin or mucosa for local effect.
- Intrathecal / epidural: injected directly into the cerebrospinal fluid or epidural space, used for spinal anaesthesia and selected chemotherapy.
Bioavailability
Bioavailability, denoted F, is the fraction of an administered dose that reaches the systemic circulation in an unchanged form. By convention an intravenous dose has a bioavailability of 1 (or 100%), and bioavailability for any other route is expressed relative to IV.
Several factors reduce oral bioavailability:
- Formulation: modified-release tablets, enteric coatings and the presence of excipients all alter dissolution.
- Gastric emptying and pH: affected by food, antacids and drugs that slow gastric motility.
- Food interactions: chelation of tetracyclines and quinolones with calcium and iron is a classic example.
- Disease of the gut: vomiting, malabsorption (Crohn's, coeliac disease, short-bowel syndrome) and gut wall oedema in heart failure all reduce absorption.
- First-pass metabolism: see below.
Diagram: Plasma concentration-time curves following an intravenous and an oral dose of the same drug. Bioavailability (F) is the ratio of the oral area under the curve (AUC) to the intravenous AUC.
First-Pass Metabolism
Drugs absorbed from the small intestine are carried in the hepatic portal vein to the liver before they reach the systemic circulation. The liver is the body's main metabolic organ and may extract and inactivate a substantial fraction of an oral dose on its very first pass. The gut wall itself also contributes some metabolism, particularly via CYP3A4 and the efflux pump P-glycoprotein.
Drugs with extensive first-pass metabolism include:
- Glyceryl trinitrate (GTN): virtually all of an oral dose is destroyed by first pass, which is why it is given sublingually or transdermally.
- Propranolol: oral bioavailability is around 25%.
- Morphine: oral bioavailability is around 30%, which is why oral and parenteral doses differ by roughly threefold.
- Lidocaine: first-pass metabolism is so extensive that lidocaine is not given orally for any therapeutic indication.
Routes that bypass first pass include intravenous, intramuscular, subcutaneous, inhaled, sublingual, buccal, transdermal and (largely) rectal administration.
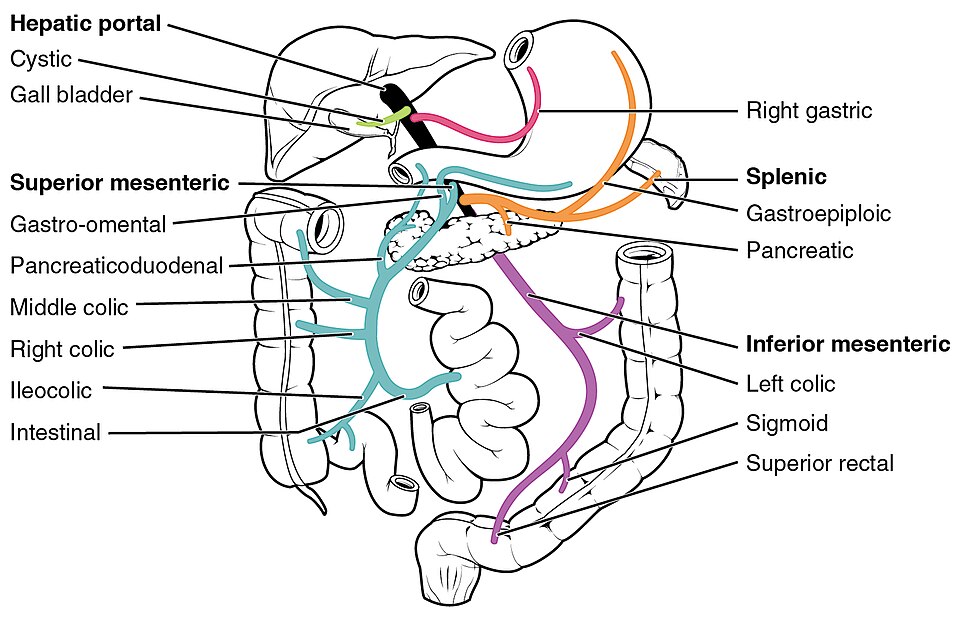
Diagram: The hepatic portal vein system. Orally absorbed drugs are carried via the portal circulation to the liver, where first-pass metabolism may inactivate a large fraction of the dose before it reaches the systemic circulation.
Creative commons source by OpenStax College, CC BY 3.0.
Distribution
Once a drug reaches the systemic circulation, it is distributed around the body. Distribution is governed by three things: how well the drug crosses capillary walls, how readily it dissolves in different tissues, and how much of it is bound to plasma proteins.
A few general rules:
- Lipophilic drugs cross cell membranes easily and reach the central nervous system, adipose tissue and other deep compartments.
- Hydrophilic drugs stay in the extracellular fluid and need junctions or transporters to leave the bloodstream.
- Highly perfused tissues (brain, liver, kidneys, heart) reach equilibrium with the blood quickly; poorly perfused tissues (fat, bone) take much longer.
Volume of Distribution
The volume of distribution (Vd) is a theoretical volume; it is the volume of fluid into which a drug would have to be dissolved to give the plasma concentration that is actually measured. The relationship is:
Vd = Dose ÷ C0 (C0 = plasma concentration immediately after an IV bolus)
Vd is best understood by reference to a few benchmark numbers in a 70 kg adult:
- Plasma volume is around 3 L.
- Extracellular fluid is around 14 L.
- Total body water is around 42 L.
A drug with a Vd close to plasma volume (around 3 L) is essentially confined to the bloodstream; heparin is a good example, because as a large polar molecule it cannot leave the vasculature. A drug with a low Vd only slightly above plasma volume is one that distributes a little further but is held in the circulation by tight protein binding; warfarin, with a Vd of around 8-10 L, falls into this group. At the other extreme, a drug with a Vd much greater than total body water has been sequestered into tissue compartments; most often fat, muscle or membrane phospholipids. Digoxin has a Vd of around 500 L, and amiodarone even more.
A useful clinical implication: the larger the Vd, the larger the loading dose required to achieve a given plasma concentration, and the longer the drug will linger after the last dose.
Plasma Protein Binding
Many drugs travel in the blood reversibly bound to plasma proteins, principally albumin (which binds acidic and neutral drugs) and α1-acid glycoprotein (which binds basic drugs). The structural reasons these proteins are such effective drug carriers are covered in Structure of Proteins. Only the free, unbound fraction is pharmacologically active and available for metabolism and excretion.
Protein binding becomes clinically important when three conditions coincide:
- The drug is highly protein bound (greater than 90%).
- The drug has a narrow therapeutic index.
- The drug has a low volume of distribution.
Warfarin, phenytoin and sulphonylureas are classic examples. If a second highly protein-bound drug displaces the first from albumin, the free fraction rises and a previously stable dose can become toxic. This is the textbook explanation for the warfarin-NSAID interaction, although in practice changes in metabolism (CYP2C9 inhibition) usually matter more.
States that reduce albumin: pregnancy, nephrotic syndrome, malnutrition, advanced liver disease and severe burns; increase the free fraction of bound drugs and predispose to toxicity at apparently normal total drug concentrations.
Metabolism
Most drugs are lipid-soluble enough to enter cells but, by the same token, lipid-soluble enough that the kidneys would simply reabsorb them from the renal tubule. Metabolism is the process of converting them to more water-soluble forms that the kidneys can excrete. The liver is by far the most important metabolic organ, although the gut wall, lung and kidneys all make a contribution. The general principles of how enzymes are regulated are covered in Enzymes and Regulation of Protein Function.
Metabolism is conventionally divided into Phase I and Phase II reactions. Most drugs undergo Phase I followed by Phase II, but either may occur alone, and a small number of drugs are excreted unchanged.
Phase I Reactions
Phase I reactions introduce or unmask a polar functional group on the drug molecule, usually by oxidation, reduction or hydrolysis. The product is a metabolite that is more water-soluble and, in most cases, less pharmacologically active than the parent drug.
There are three useful exceptions to remember:
- Pro-drugs are inactive until metabolised, for example, codeine is converted to morphine, levodopa is converted to dopamine, and enalapril is converted to enalaprilat.
- Some drugs have active metabolites that contribute to or prolong the effect of the parent drug, for example, diazepam to nordiazepam.
- Some drugs are converted to toxic metabolites, for example, paracetamol to NAPQI, which is responsible for hepatotoxicity in overdose.
Phase II Reactions
Phase II reactions conjugate the drug or its Phase I metabolite with an endogenous molecule, producing a highly water-soluble compound that is readily excreted in bile or urine. The main reactions are:
- Glucuronidation (the commonest, catalysed by UGT enzymes)
- Sulphation
- Acetylation (with genetic variation between fast and slow acetylators: relevant to isoniazid and hydralazine)
- Methylation
- Glutathione conjugation (the pathway that mops up NAPQI in paracetamol metabolism, until it is overwhelmed in overdose)
The Cytochrome P450 System
The cytochrome P450 (CYP) family is responsible for the great majority of Phase I oxidations. There are over 50 CYP enzymes in humans, but a handful do most of the drug-metabolism work, and three in particular dominate the curriculum:
- CYP3A4: metabolises around half of all licensed drugs, including statins, calcium channel blockers, macrolides and many opioids. It is also expressed in the gut wall, where it contributes to first-pass metabolism.
- CYP2D6: metabolises tricyclic antidepressants, many SSRIs, beta-blockers, codeine and tamoxifen. Its activity varies markedly between individuals: around 7% of Caucasians are poor metabolisers with a non-functional gene, while a small proportion of people from Mediterranean and East African backgrounds are ultra-rapid metabolisers who carry duplicated copies. Ultra-rapid metabolisers can develop morphine toxicity from standard codeine doses.
- CYP2C9: metabolises warfarin, phenytoin and many NSAIDs.
Genetic variation in CYP enzymes is a topic in its own right and is covered more fully in the SimpleMed article on pharmacovigilance and pharmacogenetics.
Enzyme Induction and Inhibition
CYP enzymes can be induced (their expression is upregulated, increasing metabolism) or inhibited (their activity is blocked, reducing metabolism). The clinical consequences are opposite: inducers reduce the plasma concentration of co-administered drugs and risk loss of efficacy, while inhibitors raise plasma concentrations and risk toxicity.
A useful memory aid for the common CYP inducers is PC BRAS:
- P: Phenytoin
- C: Carbamazepine
- B: Barbiturates
- R: Rifampicin
- A: Alcohol (chronic use)
- S: Sulphonylureas, St John's wort, Smoking
A matching aid for the common CYP inhibitors is AO DEVICES:
- A: Allopurinol, Amiodarone
- O: Omeprazole
- D: Disulfiram
- E: Erythromycin (and other macrolides)
- V: Valproate
- I: Isoniazid
- C: Ciprofloxacin, Cimetidine
- E: Ethanol (acute use)
- S: Sulphonamides, Some SSRIs (fluoxetine, paroxetine), Grapefruit juice
The grapefruit juice example is worth singling out: a single glass irreversibly inhibits intestinal CYP3A4 for between 24 and 72 hours, and can substantially raise plasma levels of simvastatin, ciclosporin, felodipine and several others. (The interaction with amlodipine is much weaker than with felodipine, despite both being dihydropyridines.)
Induction takes days to weeks to develop because new enzyme has to be synthesised; inhibition is usually immediate.
Excretion
Excretion is the irreversible removal of drug from the body. The kidneys do most of the work, with the biliary tract a distant second.
Renal Excretion
The kidneys receive around 25% of cardiac output, and three processes determine how much drug ends up in the urine. The underlying renal physiology is covered in The Nephron and Glomerular Filtration Rate and the Measurement of Kidney Function.
- Glomerular filtration: passive, depending on free drug concentration and GFR. Only the unbound fraction is filtered.
- Active tubular secretion: proximal tubular transporters move drug from the peritubular capillaries into the tubular lumen. This is how penicillins, methotrexate and furosemide are largely cleared, and is why probenecid (which competes for the same transporter) prolongs penicillin action.
- Passive tubular reabsorption: lipophilic drugs are reabsorbed back into the blood from the tubular fluid. Reabsorption depends on urinary pH, because only the un-ionised form crosses the membrane: alkalinising the urine traps weak acids such as aspirin in the lumen and accelerates their excretion, which is the basis of urinary alkalinisation in salicylate overdose.
Because so many drugs are renally cleared, estimating renal function is essential before prescribing. The eGFR is calculated routinely on biochemistry reports, but for drugs with a narrow therapeutic index (digoxin, gentamicin, vancomycin) the BNF still recommends using the Cockcroft-Gault formula, which is based on creatinine clearance. Pharmacokinetics also matters in Acute Kidney Injury and Chronic Kidney Disease, where falling clearance prolongs the half-life of renally excreted drugs and predisposes to toxicity.
Other Routes of Excretion
- Biliary excretion is important for large, lipophilic drugs and their conjugates. Some drugs undergo enterohepatic recirculation, in which gut bacteria deconjugate the drug and the parent compound is reabsorbed from the small intestine; this prolongs the action of, for example, the combined oral contraceptive pill, and explains the historical concern about broad-spectrum antibiotic interactions.
- Pulmonary excretion is the major route for volatile anaesthetics and ethanol (the basis of the breathalyser). The metabolic pathway for ethanol itself is described in Alcohol Metabolism.
- Smaller amounts are excreted in sweat, saliva, tears, breast milk and genital secretions.
Clearance, Half-Life and Steady State
Up to this point, ADME has been described qualitatively. To prescribe sensibly, two quantitative parameters are needed: clearance and half-life. Both follow from the volume of distribution and an understanding of elimination kinetics.
Clearance
Clearance (Cl) is the volume of plasma from which a drug is completely removed per unit time, usually quoted in millilitres per minute. It is the sum of clearance by every route; predominantly renal and hepatic, and is the parameter that, together with the desired plasma concentration, determines the maintenance dose rate:
Maintenance dose rate = Cl × Cp (target) ÷ F
A halving of clearance, whether through renal impairment, hepatic impairment or a drug interaction, doubles the steady-state plasma concentration for any given dose rate.
Half-Life
The half-life (t½) is the time taken for the plasma concentration of a drug to fall by half. For drugs that obey first-order kinetics, t½ is constant and is related to Vd and Cl by:
t½ = 0.693 × Vd ÷ Cl (0.693 = ln 2)
The take-home message is that half-life is increased either when clearance falls or when volume of distribution rises. In renal failure, falling clearance prolongs the half-life of digoxin and gentamicin; in obesity, expanded adipose tissue increases the Vd of lipophilic drugs such as diazepam and amiodarone.
Half-lives across the formulary span an enormous range. A few benchmarks worth committing to memory:
- Adenosine: under 10 seconds (used as an IV bolus in supraventricular tachycardia).
- Dobutamine: around 2 minutes (used as an IV infusion in critical care).
- Paracetamol: around 2 hours.
- Gentamicin: around 2.5 hours.
- Digoxin: around 40 hours.
- Amiodarone: several weeks.
First-Order and Zero-Order Kinetics
Most drugs at therapeutic doses are eliminated by first-order kinetics, in which the rate of elimination is proportional to the plasma concentration. A constant fraction of the drug present is removed per unit time, and the half-life is constant regardless of starting concentration. On a graph of concentration against time, first-order elimination produces an exponential decay.
A small but clinically important group of drugs follow zero-order kinetics, in which a constant amount (rather than fraction) is eliminated per unit time. This happens when the elimination machinery; typically a metabolic enzyme; is saturated. The half-life is no longer constant and a small dose increase can cause a disproportionately large rise in plasma concentration. The classic examples are remembered as PEA:
- Phenytoin
- Ethanol
- Aspirin (in overdose)
Heparin and theophylline also show zero-order kinetics at high doses. The implication for prescribing is that incremental dose changes in these drugs require considerable care, and that overdose is hazardous because the body cannot accelerate clearance.
Diagram: Comparison of first-order and zero-order elimination. First-order elimination shows exponential decay with a constant half-life; zero-order elimination is linear, with the half-life dependent on starting concentration.
Steady State and Loading Doses
When a drug is given at regular intervals, plasma concentration rises with each dose and falls between doses. After enough doses, the amount eliminated over each interval equals the amount given, and the plasma concentration oscillates around a stable mean. This is steady state, and it is where therapeutic effect is most predictable.
For first-order kinetics, steady state is reached after approximately five half-lives of regular dosing, and, conversely, it takes approximately five half-lives to clear a drug from steady state once dosing stops.
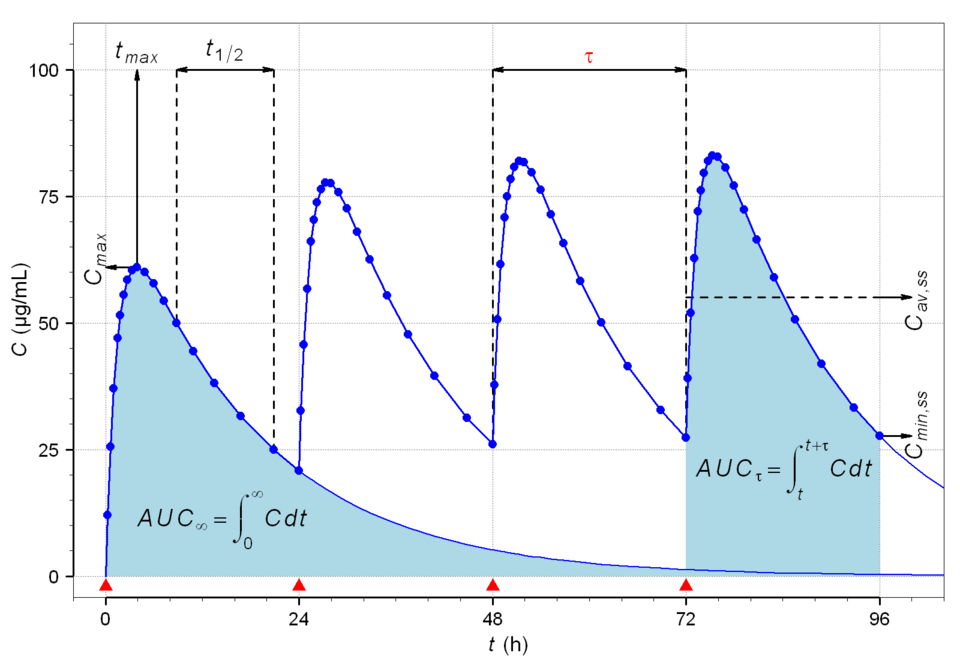
Diagram: Plasma drug concentration over 96 hours with oral dosing every 24 hours. Peak and trough concentrations rise with each dose and converge on a stable mean; the steady state; after roughly five half-lives.
Creative commons source by Helmut Schütz (Alfie66), CC BY 3.0.
Waiting five half-lives is fine for drugs with short half-lives. For drugs with long half-lives the wait becomes impractical. Digoxin, with a half-life of about 40 hours, illustrates the problem:
Worked example: digoxin in atrial fibrillation
Time to steady state = 5 × t½ = 5 × 40 hours = 200 hours ≈ 8 days
Far too slow for ventricular rate control: a loading dose is required.
A loading dose is used to fill the volume of distribution rapidly:
Loading dose = Vd × Cp (target) ÷ F
The therapeutic effect is then sustained by a maintenance dose calculated from clearance. Loading doses are routine for digoxin (in atrial fibrillation), amiodarone, phenytoin and several antibiotics including teicoplanin and vancomycin.
The Therapeutic Window
The therapeutic window is the range of plasma concentrations over which a drug is both effective and non-toxic. Drugs with a wide therapeutic window (e.g. penicillin) tolerate considerable variation in pharmacokinetics without ill effect. Drugs with a narrow therapeutic window, sometimes called narrow-therapeutic-index (NTI) drugs, do not, and small pharmacokinetic changes can push them rapidly into toxicity.
The classic NTI drugs are worth remembering, and almost all need plasma-level monitoring in routine UK practice:
- Warfarin (monitored by INR rather than direct plasma levels)
- Digoxin
- Lithium
- Phenytoin
- Theophylline
- Aminoglycosides (gentamicin, amikacin)
- Ciclosporin and tacrolimus
For all of these, anything that changes Vd, clearance or protein binding: illness, dehydration, a new co-prescription, a change in renal function; has to be considered before prescribing.
Summary
Pharmacokinetics is the framework on which safe prescribing rests. Every drug is absorbed, distributed, metabolised and excreted, and every clinical question about dosing reduces, in the end, to one of these four processes.
- Absorption sets bioavailability, with first-pass metabolism the major determinant for orally administered drugs.
- Distribution is described by the volume of distribution and modified by plasma protein binding.
- Metabolism is overwhelmingly hepatic, dominated by the cytochrome P450 system, and the most common source of clinically significant drug interactions.
- Excretion is overwhelmingly renal, which is why eGFR is the single most useful piece of information when prescribing many drugs.
- The two parameters that summarise the whole system are clearance and volume of distribution, and from them follow half-life, steady state and the loading dose.
A confident grasp of these principles makes the rest of pharmacology; drug classes, mechanisms and side effects; far easier to learn, because almost every "why?" of practical prescribing has a pharmacokinetic answer.
Reviewed by: Dr. Marcus Judge
In this article
Pharmacokinetics is the study of what the body does to a drug, summarised by the four processes of A bsorption, D istribution, M etabolism and E xcretion…
- 147

{kind=link}
{kind=link}